Física de los semiconductores orgánicos
En los semiconductores orgánicos al presentarse estructuras desordenadas y mucho menos acopladas que en los cristales atómicos que conforman a los semiconductores inorgánicos, el modelo de bandas de conducción, es de poca validez. En el caso de los cristales semiconductores, la estructura de bandas no se forma, debido a los pocos átomos que conforman cada molécula y en los polímeros semiconductores el modelo de bandas se utiliza intramolecularmente, pero intermolecularmente debido al alto grado de desorden y al bajo acople, este modelo no tiene cabida al igual que en los cristales moleculares. Por lo tanto, si mediante el modelo de bandas no se puede explicar el transporte de cargas en los semiconductores orgánicos, ¿mediante que modelo se explica dicho transporte?. Según Vissemberg[23], el fenómeno de conducción en los semiconductores orgánicos se explica de la siguiente manera: en los semiconductores orgánicos los electrones se encuentran en estados energéticos o niveles de energía localizados, a diferencia de los conductores y semiconductores tradicionales en donde los electrones están deslocalizados por todo el material. En los materiales orgánicos, estos estados localizados o sitios son: los estados de las moléculas individuales en los cristales moleculares, los estados de las cadenas polimericas individuales o los estados de los segmentos de estas cadenas donde la conjugación es interrumpida por defectos estructurales o químicos. Habiendo definido los estados localizados o sitios y teniendo presente que estos sitios actúan como pozos de potencial, la transferencia de carga entre sitios se da mediante saltos cuánticos o hopping en donde los portadores de carga mediante efecto túnel asistido por fonones (vibraciones de la estructura del material) pasan de un sitio a otro, bajo ciertas condiciones especiales. Para entender un poco más este fenómeno, vamos a explicar en forma general este mecanismo cuántico. El efecto túnel, como dice la editorial Time Life[24], fue por primera vez propuesto por George Gamow a principios del siglo XX, para explicar como dos átomos de hidrogeno se fusionaban en las estrellas a temperaturas sumamente bajas para que se diera dicho fenomeno, y se basa en el principio de incertidumbre y en la ecuación de onda de Schrödinger, la cual predice que una partícula subatómica dentro de un pozo de potencial o fuera de éste, siempre y cuando la barrera de potencial sea finita, tiene una probabilidad (aunque muy pequeña) de encontrarse por fuera o de entrar a dicho pozo, atravesando sus paredes, como si hiciera un túnel, razón por la cual se denominó efecto túnel.
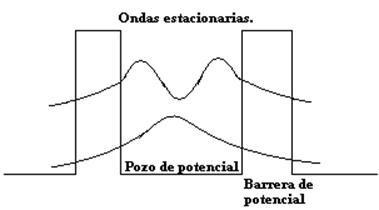
Figura 15.Pozo de potencial o sitio.
En un semiconductor orgánico, los electrones como dijimos en el capítulo anterior, se encuentran deslocalizados dentro de los sistemas π de las moléculas individuales de los cristales moleculares o de las cadenas poliméricas. "Estos electrones al ser estimulados por un campo eléctrico se mueven dentro de estos sitios de extremo a extremo y al encontrarse con una barrera energética rebotan y forman ondas estacionarias"[25] que tienen cierto nivel de energía. Teniendo en cuenta lo anterior los electrones u ondas estacionarias tienen la posibilidad de atravesar la barrera de potencial entre las moléculas o polímeros, siempre y cuando, según Richard Turton[26], los electrones de un sitio origen esten en un nivel de energía igual a uno de los niveles de energía permitido en el sitio al que va a trasladarse, de lo contrario el hopping no se lleva a cabo. Por lo tanto, con el fin de lograr los niveles de energía necesarios, el papel de los fonones es indispensable, ya que la energía de los fonones es trasmitida a los electrones que de esta forma alcanzan la energía suficiente para trasladarse por efecto túnel.
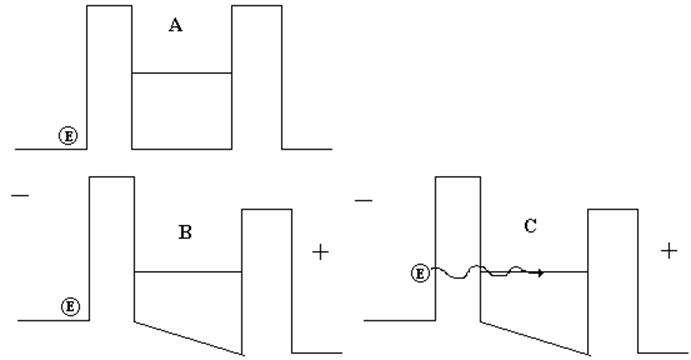
Figura 16. Hopping asistido por fonones.
En la figura 16, en la parte A, observamos al lado izquierdo un electrón en una molécula o sitio con una energía inferior a la energía de uno de los niveles de energía discretos de otra molécula o sitio, inhabilitando esto su traslado por efecto túnel. En la parte B, al aplicarle un campo eléctrico al material los niveles de energía al igual que en los semiconductores inorgánicos se inclinan y la diferencia de energía entre los dos sitios disminuye pero aun no coinciden. Sin embargo en la parte C, el electrón absorbe la energía de un fonón e iguala la energía de la molécula vecina, situación ésta que le permite trasladarse por hopping.
"El mecanismo de efecto túnel asistido por fonones o Hopping (saltos cuánticos) fue originalmente propuesto por Conwell y Mott en los semiconductores inorgánicos con defectos trampas y la tasa de transición por efecto túnel asistido por fonones fue calculada por Miller y Abrahams, en donde el Hopping desde un estado localizado J a un estado i se lleva a cabo a una frecuencia para el fonón de νo, la cual es adecuada para que el fonón sea absorbido por los portadores de carga, que así aumentan su energía y se trasladan por efecto túnel"[27].
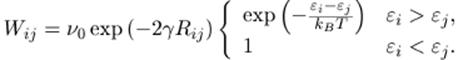
Ecuación 1. Tasa de tunelage asistido por fonones.
En la ecuación 1, γ es la longitud inversa de localización, Rij es la distancia entre los estados localizados, εi es la energía en el estado i y εj es la energía del estado J. Como los índices de salto son fuertemente dependientes de la energía y la posición de los estados localizados, el transporte por Hopping es extremadamente sensible, tanto a la estructura como al desorden energético. La anterior es la razón por la que los cristales moleculares tienen una conductividad mayor que los polímeros semiconductores.
Conociendo ya el modelo mediante el cual se explica la semiconducción en los materiales orgánicos, es importante conocer, como es la movilidad en estos materiales.
2.1 MOVILIDAD EN LOS SEMICONDUCTORES ORGÁNICOS.
Para medir esta característica en los semiconductores inorgánicos, los investigadores utilizan la técnica del tiempo de vuelo, la cual consiste en "generar mediante un flash de luz, una capa de portadores de carga cerca de un electrodo de un capacitor de placas paralelas y bajo un campo eléctrico aplicado E, hacer que las cargas se mueven hacia el electrodo opuesto. El tiempo de tránsito τt al cual las cargas alcanzan ese electrodo (distancia L) es entonces una medida directa de la movilidad de los portadores"[28].
Ecuación 2. Movilidad por la técnica de tiempo de vuelo.
Sin embargo en los semiconductores orgánicos según Vissenberg[29], las foto-corrientes generadas son con frecuencia dispersivas (disminuyen con el tiempo debido a que las cargas toman diferentes caminos y se inmovilizan temporalmente debido a que quedan atrapadas en irregularidades) y por lo tanto la velocidad de las cargas disminuye cuando la muestra es atravesada. En ese caso el tiempo de arribo promedio de las cargas y consecuentemente la movilidad de los portadores, dependen de la dimensión de la muestra y la movilidad no refleja un parámetro genuino del material. Esa dispersión de la corriente es debido, a que como vimos en el capítulo anterior, los semiconductores orgánicos presentan bajo acople y en especial los polímeros tienen un grado muy alto de desorden al ser amorfos. Teniendo en cuenta lo anterior y según Man Hoy Wong[30], investigaciones con respecto a la movilidad, han concluido que la movilidad en los materiales orgánicos tiene una dependencia del campo eléctrico µ = µ0 exp(β√E) que es del tipo Poole-Frenkel y adicionalmente se observa un transporte de carga tanto dispersivo como no dispersivo. Por lo tanto, para estudiar la movilidad en los semiconductores orgánicos se utiliza el modelo de Bässler, el cual modela ambos comportamientos, teniendo en cuenta la simulación de Monte Carlo, la cual se basa en una distribución Gaussiana de densidad de estados (DOS) o niveles de energía disponibles para los electrones, con el cual se obtienen unos resultados muy acordes con los experimentos. Dichos resultados muestran que el transporte de carga en los sólidos orgánicos es altamente dispersivo, y que la movilidad de los portadores depende de tres aspectos fundamentales que son: el desorden energético (debido a la fluctuación de la polarización de la energía de la retícula y/o de la distribución de la longitud de los enlaces del semiconductor), la intensidad del campo eléctrico aplicado y el desorden estructural (debido a la fluctuación de la distancia entre sitios, la cual varía con una varianza Σ).
Para describir los tres aspectos anteriores, el modelo de Bässler, primero calcula, para un paquete de portadores de carga, la energía media de equilibrio en función del desorden energético (<ε∞>), teniendo en cuenta que los portadores de carga no son sometidos a un campo electrico.
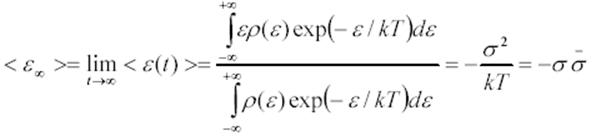
Ecuación 3. Energía media de equilibrio.
Donde ρ(ε) es la densidad de estados de energía y 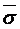 es el ancho de la DOS normalizados a kT y representa el desorden energético. Los sistemas que poseen grandes desordenes energéticos implican bajas temperaturas de acuerdo a la ecuación y se equilibran más despacio debido a que los saltos activados térmicamente, que ayudan a los portadores a encontrar otras trayectorias para la relajación, están eliminados.
es el ancho de la DOS normalizados a kT y representa el desorden energético. Los sistemas que poseen grandes desordenes energéticos implican bajas temperaturas de acuerdo a la ecuación y se equilibran más despacio debido a que los saltos activados térmicamente, que ayudan a los portadores a encontrar otras trayectorias para la relajación, están eliminados.
Según Man Hoy Wong[31], el segundo paso del modelo de Bässler, con el fin de encontrar una ecuación analítica de la movilidad, tiene en cuenta la dependencia que tienen la movilidad, de la temperatura, la cual tiene la siguiente ecuación:
Ecuación 4. Dependencia térmica de la movilidad
Luego, teniendo la relación de la movilidad con la temperatura, el modelo de Bässler relaciona la movilidad conjuntamente con el campo eléctrico y el desorden energético, y aquí encuentra un comportamiento como el de la figura 17, en donde el desorden posicional o varianza es cero (Σ = 0) y la movilidad del material "presenta un comportamiento que se aproxima a (logµ~βE) que es del tipo de Poole-Frenkel, el cual se da a campos eléctricos moderadamente altos (>10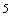 V/cm) con tendencia a saturarse a grandes campos eléctricos (>10
V/cm) con tendencia a saturarse a grandes campos eléctricos (>10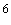 V/cm)"[32]. En la figura se ve que a medida que el desorden energético aumenta, la movilidad disminuye, lo cual es consecuencia de lo que vimos anteriormente cuando tratamos el pozo de potencial, en donde el trasporte por efecto túnel necesita que las energías de los sitios entre los cuales se dará el hopping sean iguales y el desorden energético implica que dichas energías son muy dispares. También la figura 17 nos muestra que la movilidad aumenta con el aumento del campo eléctrico, lo cual es consecuencia de que la DOS aumenta con el campo y el aumentar la densidad de estados, conlleva inevitablemente a una disminución en el desorden energético. Esto también se puede explicar retomando la figura 16, en donde observamos que un potencial aplicado reduce la barrera de potencial, facilitando el hopping en la dirección del campo aplicado. Por último y como más importante, la figura nos muestra que a grandes campos, la movilidad alcanza un nivel de saturación en el cual deja de depender del desorden energético
V/cm)"[32]. En la figura se ve que a medida que el desorden energético aumenta, la movilidad disminuye, lo cual es consecuencia de lo que vimos anteriormente cuando tratamos el pozo de potencial, en donde el trasporte por efecto túnel necesita que las energías de los sitios entre los cuales se dará el hopping sean iguales y el desorden energético implica que dichas energías son muy dispares. También la figura 17 nos muestra que la movilidad aumenta con el aumento del campo eléctrico, lo cual es consecuencia de que la DOS aumenta con el campo y el aumentar la densidad de estados, conlleva inevitablemente a una disminución en el desorden energético. Esto también se puede explicar retomando la figura 16, en donde observamos que un potencial aplicado reduce la barrera de potencial, facilitando el hopping en la dirección del campo aplicado. Por último y como más importante, la figura nos muestra que a grandes campos, la movilidad alcanza un nivel de saturación en el cual deja de depender del desorden energético
Fuente: WONG, Man Hoy. Charge transport and injection in doped organic semiconductors. Universidad de Cornell. Mayo de 2004. <my.ece.ucsb.edu/mhwong/documents/thesis.pdf> [consulta: feb 2005]
El modelo de Bässler habiendo ya considerado el desorden energético, continua su análisis, considerando la movilidad relacionada conjuntamente con el desorden posicional y el campo eléctrico y en este aspecto encuentra que la movilidad se comporta como la figura 18, en donde el desorden energético es cero. En esta figura se observa que al aumentar el desorden posicional se aumenta la movilidad, lo cual se debe a que este desorden introduce muchas rutas alternas, es decir, que si en la dirección del campo eléctrico la barrera de potencial es muy grande, es factible que en otras direcciones se encuentren barreras menos anchas por las cuales los portadores puedan tomar atajos, incluso en direcciones en contra del campo eléctrico. Sin embargo a medida que el campo aplicado aumenta, los portadores de carga son obligados a obedecer la ruta señalada por el campo y las rutas alternas ya no se pueden tomar, con lo cual disminuye la movilidad.

Fuente: WONG, Man Hoy. "Charge transport and injection in doped organic semiconductors." Universidad de Cornell. Mayo de 2004. <my.ece.ucsb.edu/mhwong/documents/thesis.pdf> [consulta: feb 2005]
Habiendo considerado los tres factores de los que depende la movilidad, el modelo de Bässler, con el objeto de encontrar una fórmula general para la movilidad, toma todos los factores en conjunto y llega una ley universal que relaciona los tres efectos:
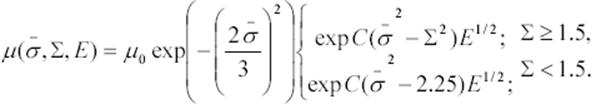
Ecuación 5. Movilidad relacionada con E y el desorden energético y posicional.
En la ecuación 5, C es un parámetro del sistema que depende del material.
Teniendo en cuenta todo lo anterior se puede concluir que:
- Según Man Hoy Wong[33], Mediante una combinación adecuada de desorden energético y de desorden posicional se puede lograr que la movilidad de un semiconductor orgánico no dependa del campo eléctrico aplicado (sea constante en la práctica), siempre y cuando este se mantenga dentro de un rango específico.
- El desorden energético disminuye la movilidad y en si mismo disminuye con el campo eléctrico.
- El desorden posicional aumenta la movilidad, pero también disminuye con el campo eléctrico.
- El campo eléctrico hace que la movilidad de un semiconductor orgánico no sea constante (pero este efecto tiene solución) y satura la movilidad de los portadores de carga a campos elevados.
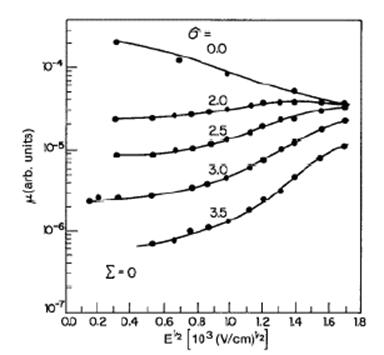
La figura 19 muestra la movilidad de los huecos en TAPC/polycarbonate ante la presencia de ambos parámetros de desorden y del campo eléctrico.
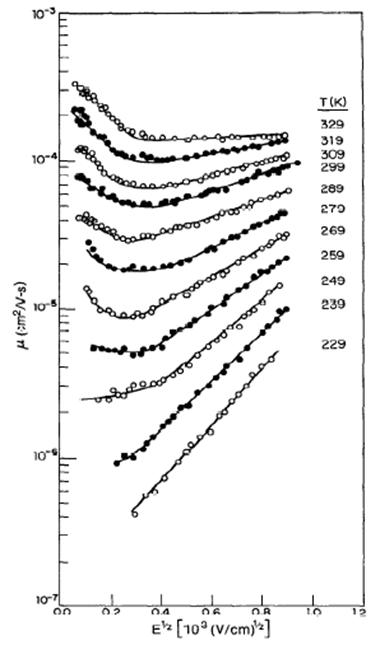
Fuente: WONG, Man Hoy. "Charge transport and injection in doped organic semiconductors." Universidad de Cornell. Mayo de 2004. <my.ece.ucsb.edu/mhwong/documents/thesis.pdf> [consulta: feb 2005]
Ider Guerrero
EES
Secc: 1
No hay comentarios:
Publicar un comentario